MetaCerberus Tutorial - What’s in the Results folder?
Now that we’ve run the MetaCerberus pipeline, let’s take a look at the results folder. Having ran the --super option on my data, the results folder looks like so:

Now a closer look at each subdirectory of our results:
Step_5-format:

Step 5 contents only consist of a complete file, which merely indicates Step 5 ran to completion.
Step_6-metaomeQC
Here are the contents of step_06-metaomeQC:
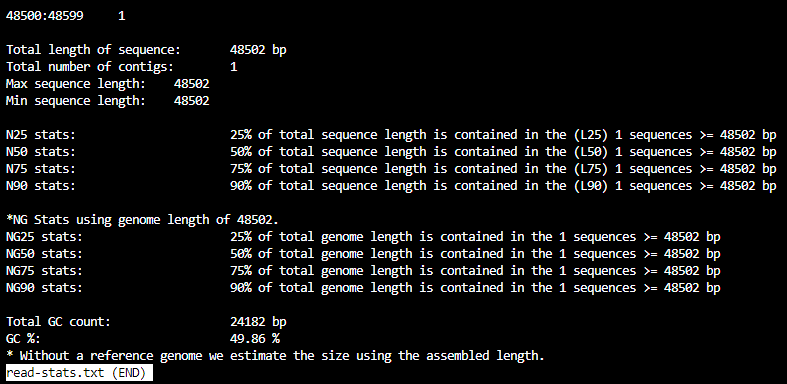
The file read-stats.txt contains statistics for your input file, like so:
Note
The file stderr.out is a log file where any error messages will be stored.
Step_7-geneCall
Contents of the step_07-geneCall directory are:
These are protein files in different formats.
Step_8-hmmer
Contents of the step_08-hmmer directory are:
For your MetaCerberus run, you should get a subdirectory for the mode that MetaCerberus used (FragGeneScan, Prodigal, Prodigalgv, etc). In this example run, we have several file outputs for FragGeneScan. The types of outputs are similar for prodigal.
This is what they look like:
Note
.tsv files can be opened with Excel.
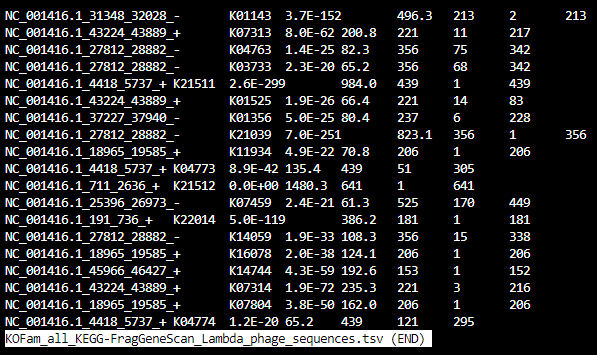
KOFam_all_FOAM-FragGeneScan_Lambda_phage_sequences.tsv:
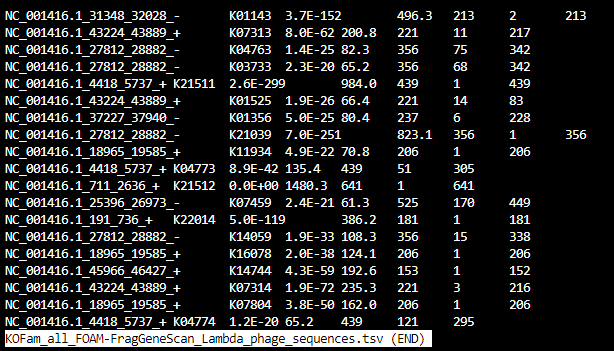
KOFam_all_KEGG-FragGeneScan_Lambda_phage_sequences.tsv
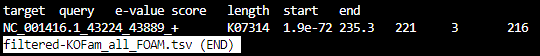
filtered-KOFam_all_FOAM.tsv
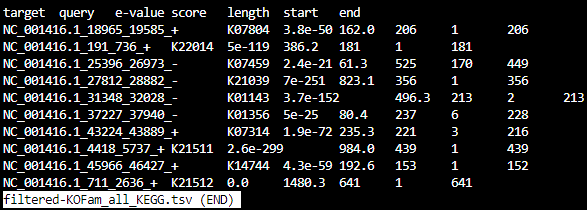
filtered-KOFam_all_KEGG.tsv
filtered.tsv
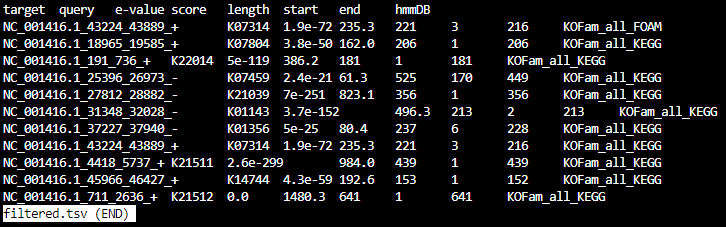
Step_9-parse
The contents of step_09-parse are:
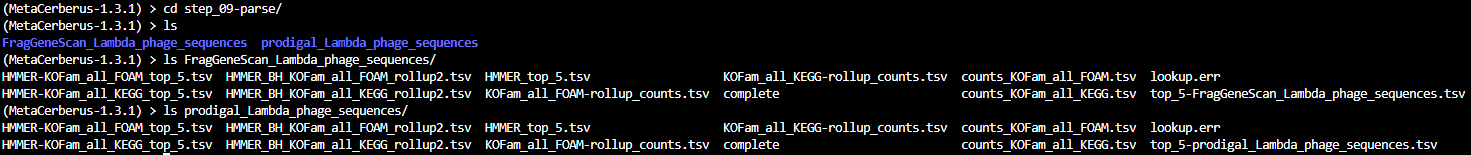
- Quick link to examples:
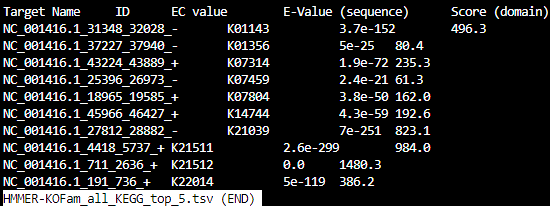
Looking a little closer:
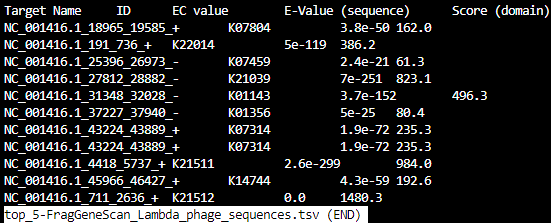
HMMER-KOFam_all_FOAM_top_5.tsv

HMMER-KOFam_all_KEGG_top_5.tsv
HMMER_BH_KOFam_all_FOAM_rollup2.tsv
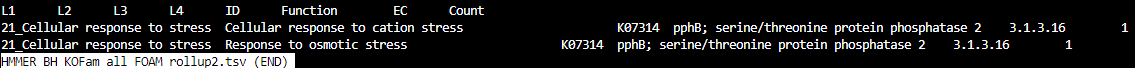
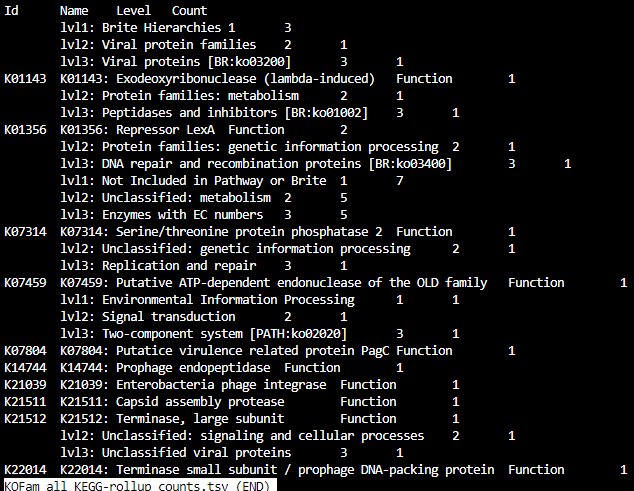
HMMER_BH_KOFam_all_KEGG_rollup2.tsv

HMMER_top_5.tsv
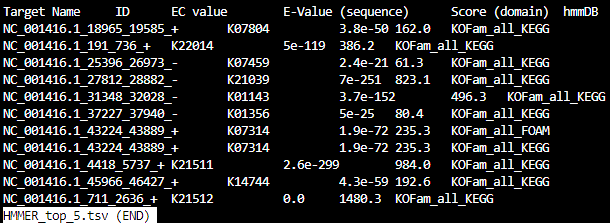
KOFam_all_FOAM-rollup_counts.tsv
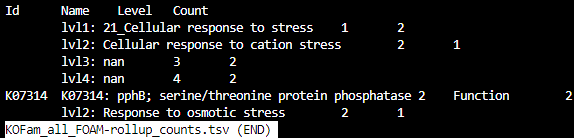
KOFam_all_KEGG-rollup_counts.tsv
counts_KOFam_all_FOAM.tsv
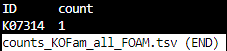
counts_KOFam_all_KEGG.tsv
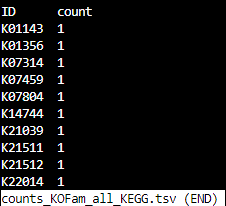
top_5-FragGeneScan_Lambda_phage_sequences.tsv
Step_10-visualizeData
The contents of step_10-visualizeData are:

What’s in the FragGeneScan and Prodigal subdirectories?

Files under FragGeneScan or Prodigal:
- Quick link to examples:
KOFam_all_FOAM_level-1.tsv

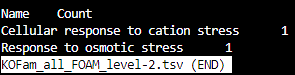
KOFam_all_FOAM_level-2.tsv
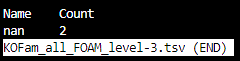
KOFam_all_FOAM_level-3.tsv
KOFam_all_FOAM_level-4.tsv
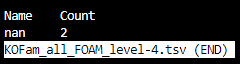
KOFam_all_FOAM_level-id.tsv

KOFam_all_KEGG_level-1.tsv

KOFam_all_KEGG_level-2.tsv
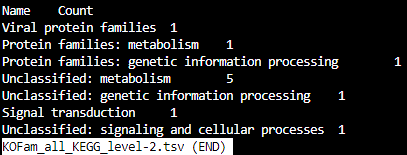
KOFam_all_KEGG_level-3.tsv
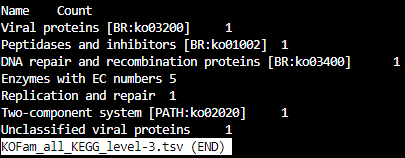
KOFam_all_KEGG_level-id.tsv

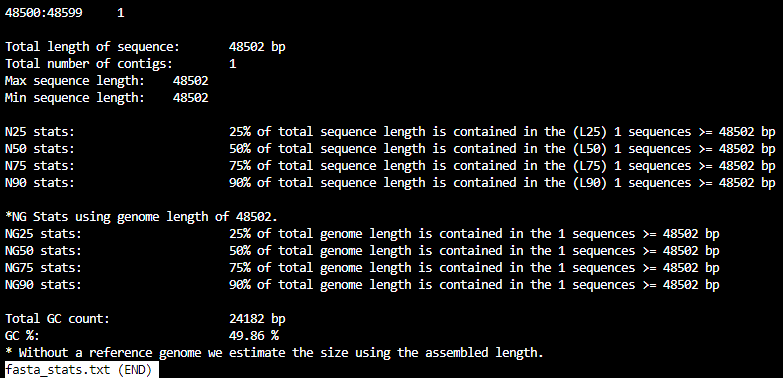
fasta_stats.txt
sunburst_KOFam_all_FOAM.html — open in web browser
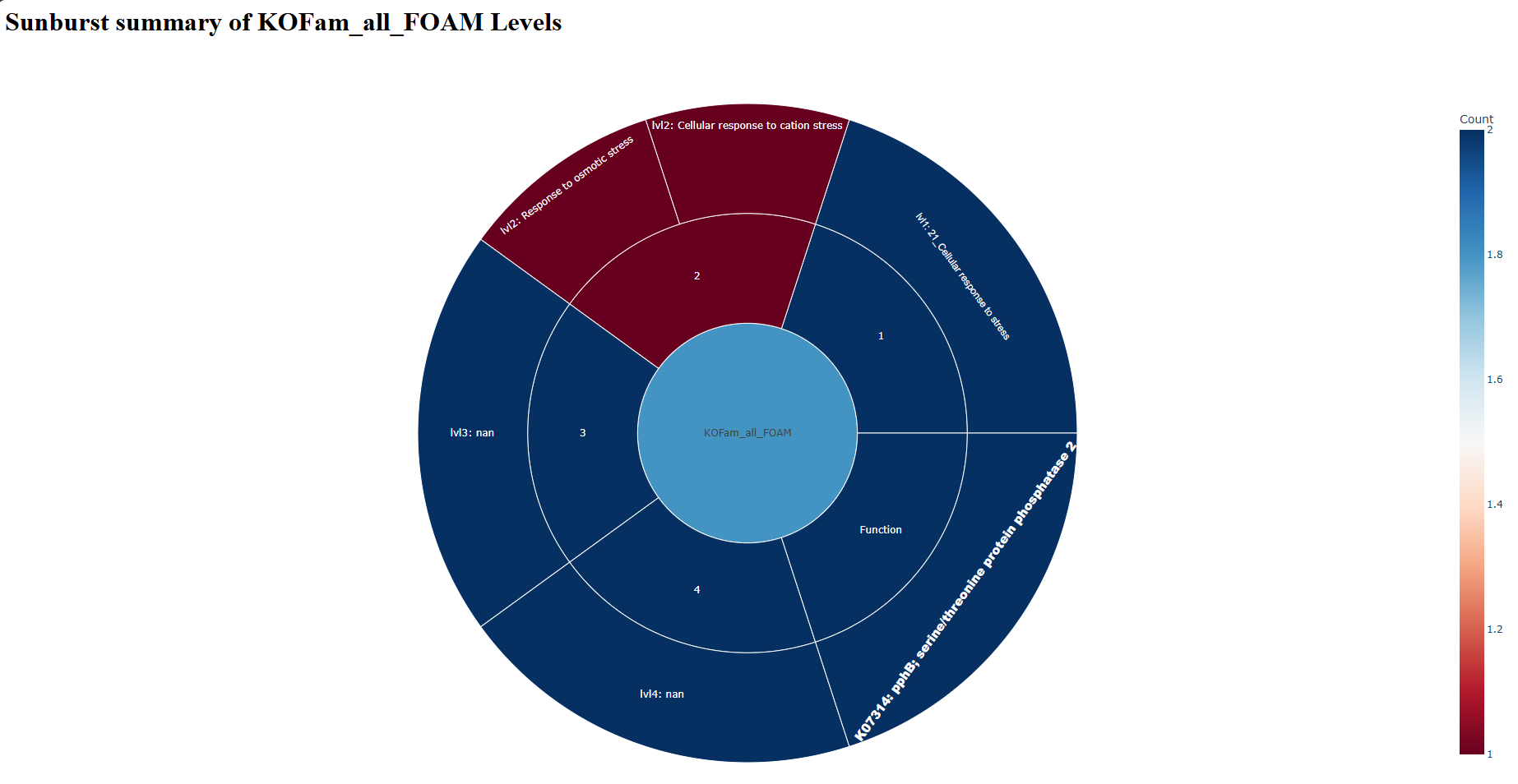
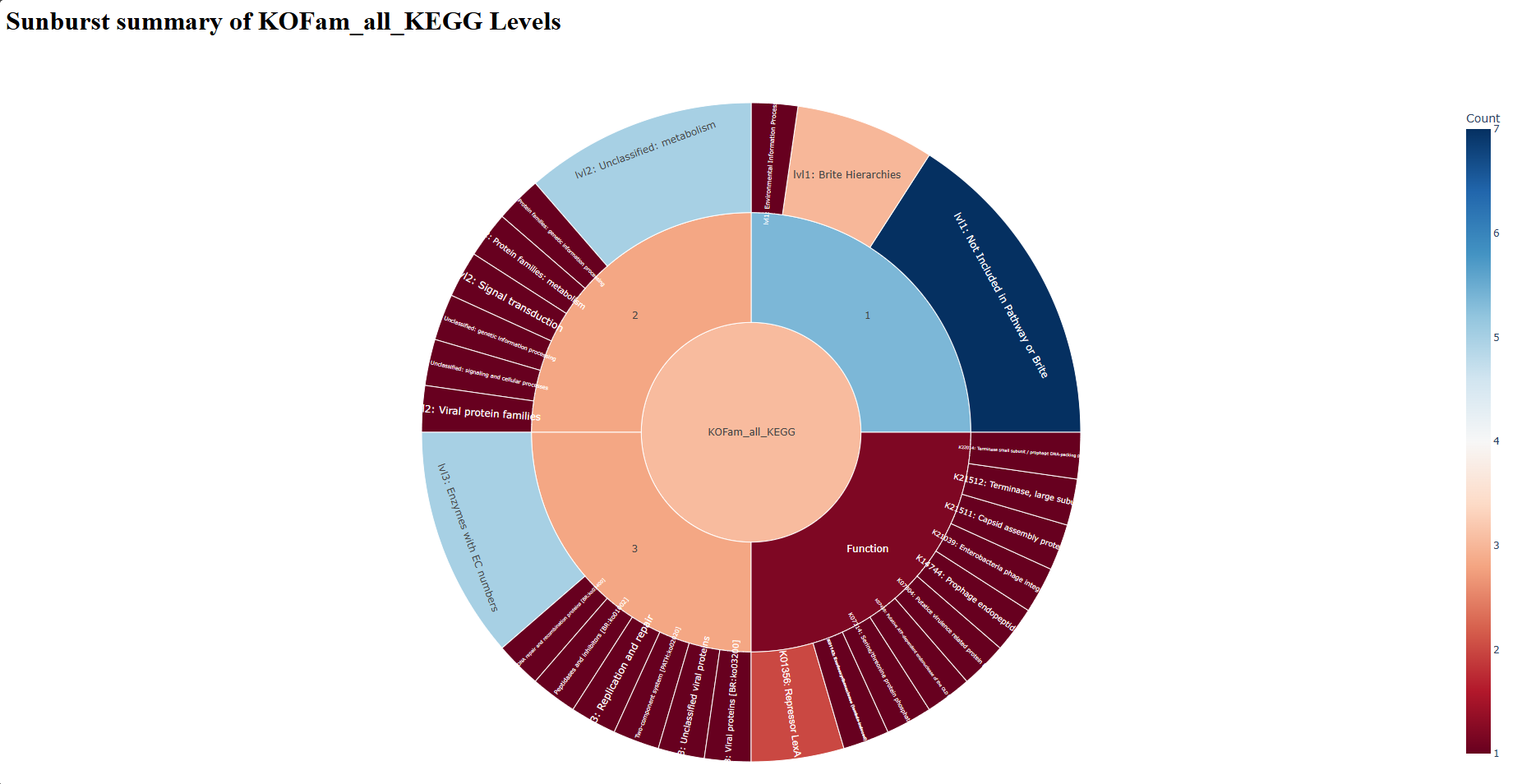
sunburst_KOFam_all_KEGG.html — open in web browser
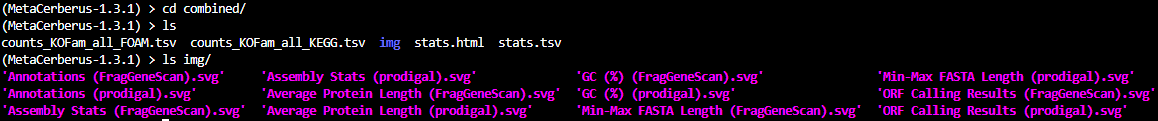
Contents under combined:
At a glance:

- Quick link to examples:
counts_KOFam_all_FOAM.tsv
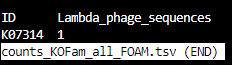
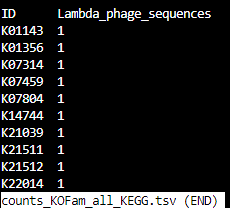
counts_KOFam_all_KEGG.tsv
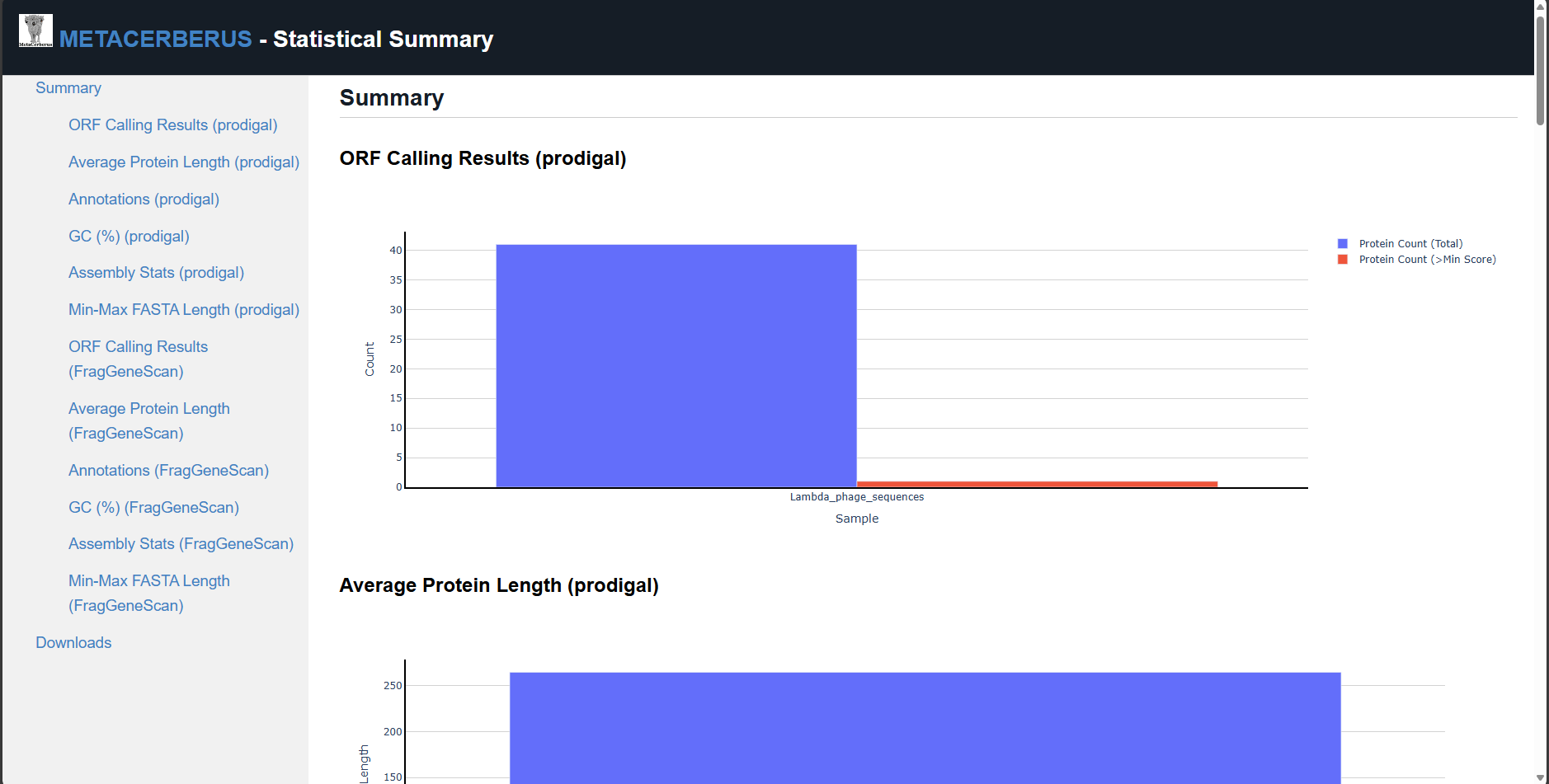
stats.html — open in web browser
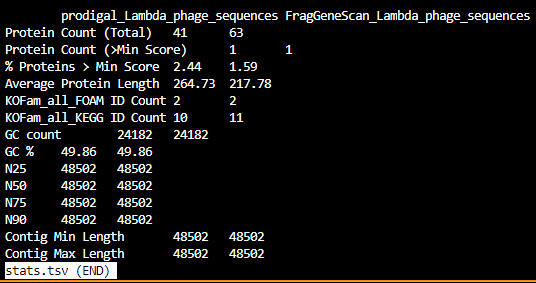
stats.tsv
img — contains the individual .png image files which are collectively located in stats.html
Final
The contents of final are:
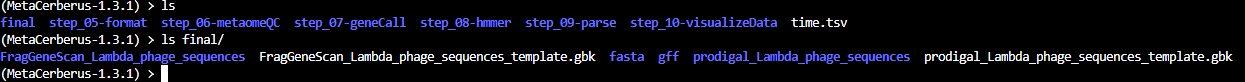
There’s two
.gbkfiles which are in GenBank Format.The
./final/fastasubdirectory contains .faa, .ffn, and .fna files of FragGeneScan, Prodigal, etc (depending on commands given):
.faa- Protein FASTA file of the translated CDS/ORFs sequences
.ffn- FASTA Feature Nucleotide file, the Nucleotide sequence of translated CDS/ORFs.
.fna- Nucleotide FASTA file of the input contig sequences.

The
./final/gffcontains .gff and .gtf files:
.gff- General Feature Format
.gtf- Gene Transfer Format
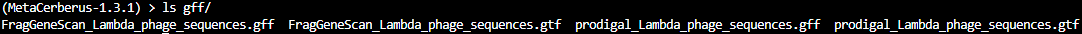
What’s in the ./final/FragGeneScan_<file_name> and ./final/prodigal_<file_name> subdirectories?

A closer look:
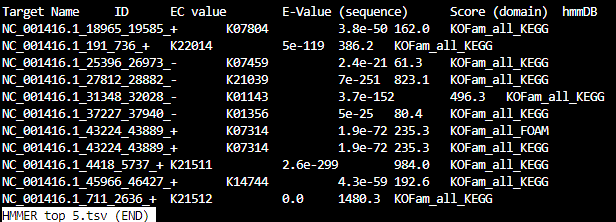
HMMER_top_5.tsv
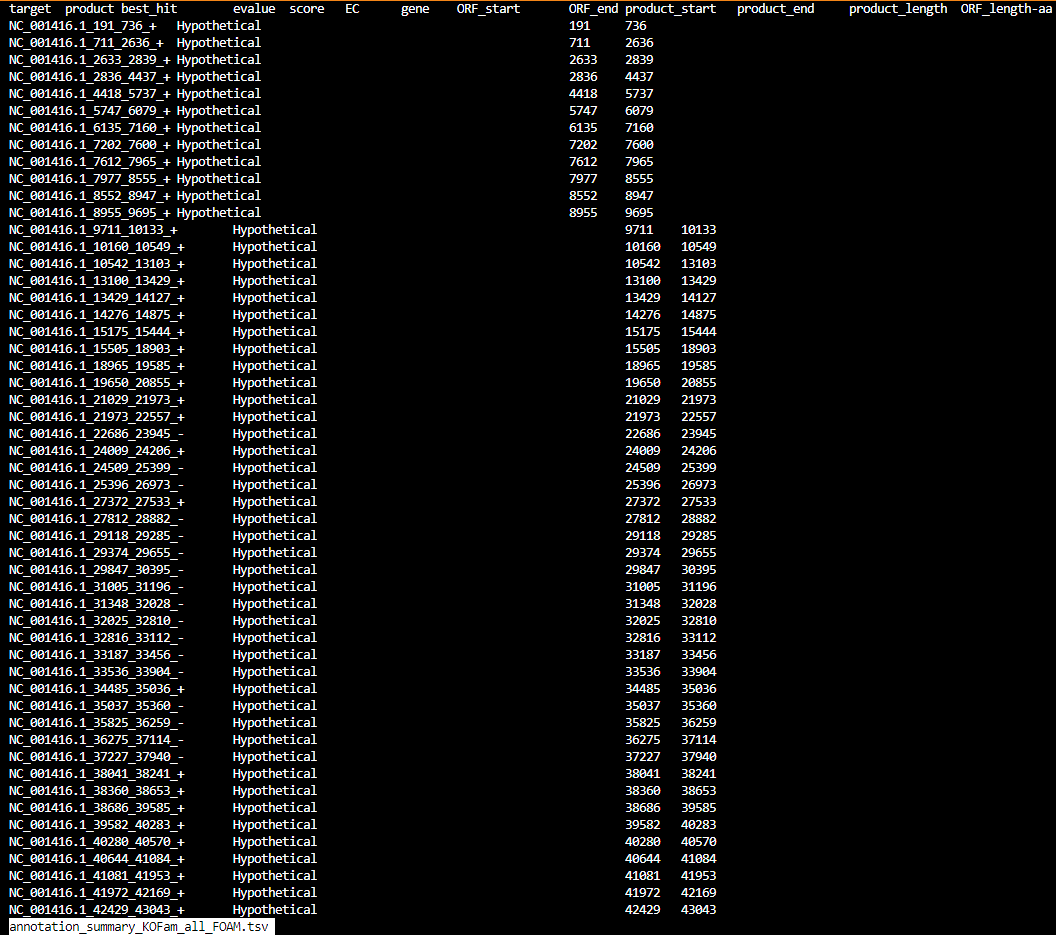
annotation_summary_KOFam_all_FOAM.tsv
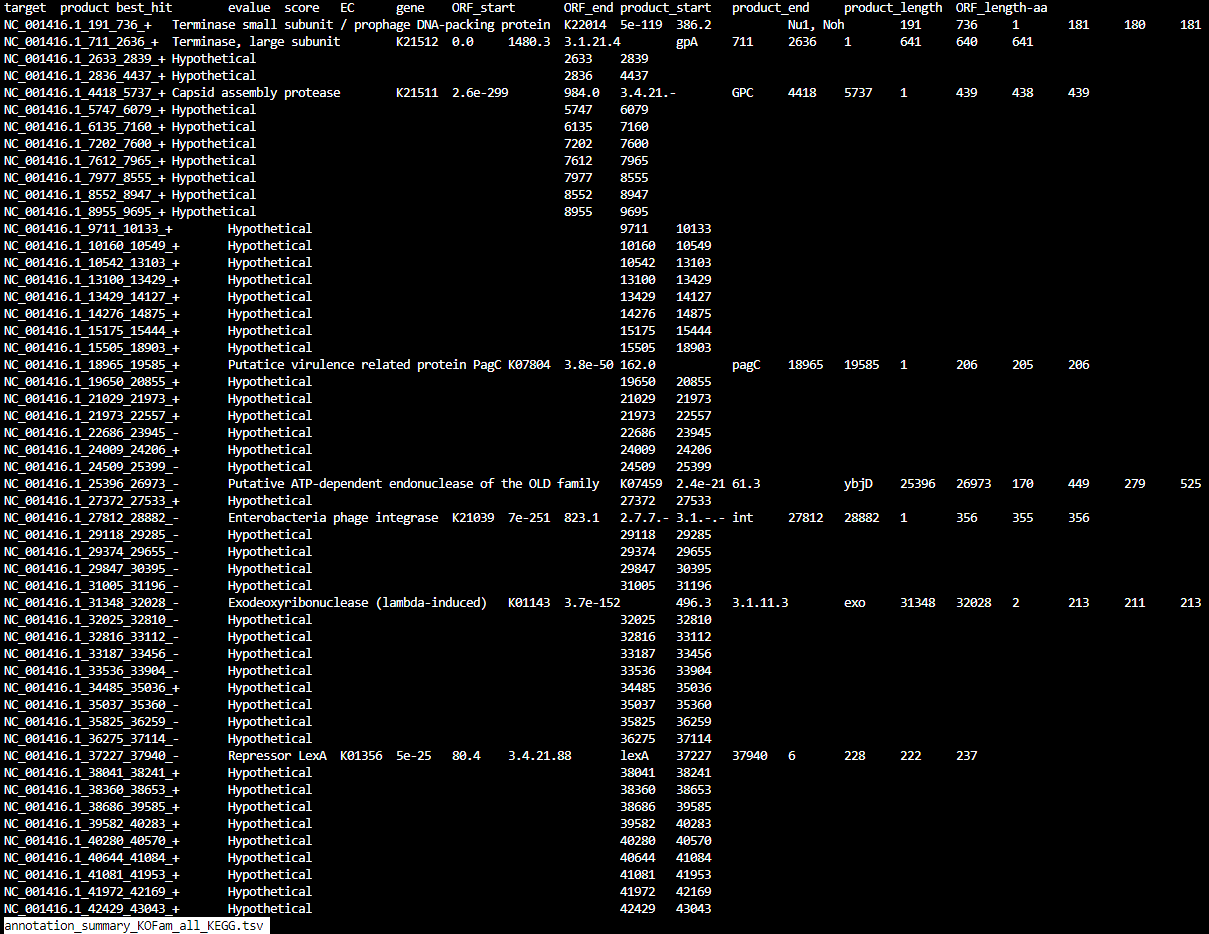
annotation_summary_KOFam_all_KEGG.tsv
final_annotation_summary.tsv

rollup_KOFam_all_FOAM.tsv
rollup_KOFam_all_KEGG.tsv
